

About OMiMa
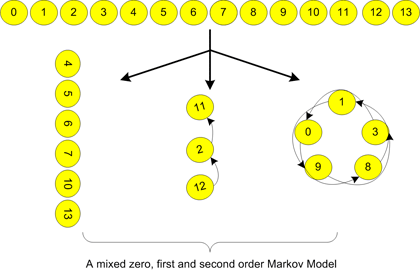
The OMiMa System is a computational tool for identifying functional motifs in DNA or protein sequences. OMiMa System is based on the Optimized Mixture of Markov models that are able to incorporate most dependencies within a motif. Most important, OMiMa is capable to adjust model complexity according to motif dependency structures, so it can minimize model complexity without compromising prediction accuracy. OMiMa uses our fast Markov chain optimization method, the Directed Neighbor-Joining (DNJ), which makes OMiMa more computationally efficient.
Availability and Citation
OMiMa is freely available to public and can be downloaded at the following links.
- OMiMa for Linux (666KB)
- OMiMa for 64-bit Linux (1MB)
Please use the commands 'tar xvfz *.tar.gz' to uncompress the downloaded file, then follow the instructions in the OMiMa instructions for usage.
OMiMa should be cited as
Weichun Huang, David M Umbach, Uwe Ohler, Leping Li. Optimized mixed Markov models for motif identification.
Test Data and Results
The two original donor splice datasets were from Reese (the small set), and from Yeo and Burge (the large set). TFBS simulated data and the reformatted training and testing datasets for OMiMa can be downloaded at the following links.
- Simulation data of palindromic motifs (5KB) - [Read Me First (1KB)]
- Small dataset from Reese [donorSS_Reese.tar.gz] (5KB) - [Read Me First (824B)]
- Large dataset from Yeo and Burge [donorSS_Yeo.tar.gz] (2MB) - [Read Me First (802B)]
- 5 training subsets sampled from Yeo and Burge's original training dataset [trainset60Pct.tar.gz] (1MB) - [Read Me First (627B)]
- OMiMa's prediction results based on each of the above 5 training subsets of Yeo's data
Note: some above files for download are in *.tar.gz format (use command 'tar xfz afile.tar.gz' to extract files). If you would like to have other data or programs used in our paper, please feel free to contact me.