

Mitochondrial Genetic Diseases

William C. Copeland, Ph.D.
[email protected]
Research Summary
William C. Copeland, Ph.D., is Chief of the Genome Integrity and Structural Biology Laboratory and head of the Mitochondrial DNA Replication Group, which studies mitochondrial diseases.
Mitochondrial diseases are devastating disorders for which there is no cure and no proven treatment. About 1 in 4000 individuals is at risk of developing a mitochondrial disease sometime in their lifetime. Half of those affected are children who show symptoms before age five, and approximately 80% of them will die before age 20. The mortality rate is roughly that of cancer. The human suffering imposed by mitochondrial and metabolic diseases is enormous, yet much work is needed to understand the genetic and environmental causes of these diseases. Mitochondrial genetic diseases are characterized by alterations in the mitochondrial genome, as point mutations, deletions, rearrangements, or depletion of the mitochondrial DNA (mtDNA). The mutation rate of the mitochondrial genome is 10–20 times greater than of nuclear DNA, and mtDNA is more prone to oxidative damage than is nuclear DNA. Mutations in human mtDNA cause premature aging, severe neuromuscular pathologies and maternally inherited metabolic diseases, and influence apoptosis.
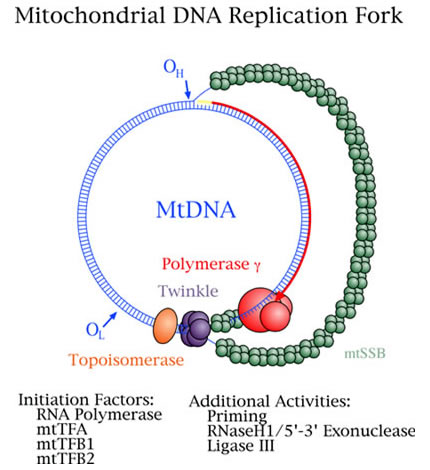
The primary goal of the Mitochondrial DNA Replication Group is to understand the role of the replication apparatus in the production and prevention of mutations in mtDNA. Because the genetic stability of mtDNA depends on the accuracy of DNA polymerase gamma (pol γ), this project focuses on understanding the role of human pol γ in mtDNA mutagenesis. Mutations in the POLG gene for the catalytic subunit of pol γ have been linked to several mitochondrial disorders, including progressive external ophthalmoplegia, sensory and ataxic neuropathy, Alpers syndrome, and male infertility. To date, nearly 90 disease mutations and nine non-synonymous polymorphisms have been identified in the POLG coding region. The group is studying the molecular effects of disease mutations in pol γ and has developed a web-based database that lists all published mutations in the POLG coding region and describes the associated diseases.
Major areas of research:
- Mitochondrial diseases and mitochondrial DNA replication
- Disease mutations in the gene for DNA pol γ
- Mitochondrial toxicity induced by anti-HIV nucleoside analogs
Current projects:
- Characterization of the role of human pol γ in mtDNA mutagenesis
- Study of the molecular effects of disease mutations in pol γ and development of a web-based database that lists all published mutations in the POLG coding region and describes the associated diseases
- Elucidation of the role of the human pol γ in induced mitochondrial toxicity caused by anti-HIV nucleoside analogs
Copeland received his Ph.D. in chemistry/biochemistry from the University of Texas at Austin in 1988. He completed his postdoctoral training at Stanford University School of Medicine, Department of Pathology studying the human DNA polymerase alpha/primase complex. He has published nearly 70 articles in peer-reviewed journals. Copeland served as president of the Mitochondrion Research Society from 2005 to 2007 and is currently the chair of the grants committee for the United Mitochondrial Disease Foundation. He joined NIEHS in 1993.