

Environmental Cardiopulmonary Disease Group
The basis of the Zeldin group's work in this area is the hypothesis that cyclooxygenase (COX)-derived eicosanoids (Figure 1) are important modulators of the lung immune response to environmental agents (Figure 2).
The group was the first to show that COX-1 is the predominant enzyme that biosynthesizes prostaglandin E2 in the lung (Gavett et al., J. Clin. Invest., 1999).
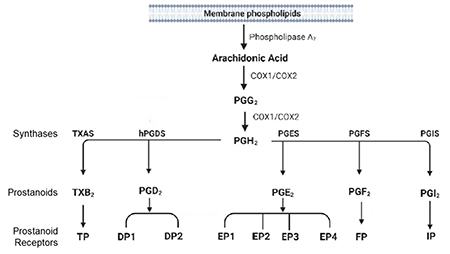
Figure 1. Metabolism of Arachidonic Acid by Cyclooxygenases and Prostaglandin Synthases. Arachidonic acid can be metabolized by cyclooxygenases (COX-1 and COX-2) to prostaglandin H2 (PGH2) that is further metabolized by secondary synthases to form specific prostanoids such as PGE2, PGD2, PGF2α, prostacyclin (PGI2) and thromboxane (TXA2). These prostanoids can signal through G-protein coupled receptors, including those from the EP, DP, FP, IP and TP subfamilies.
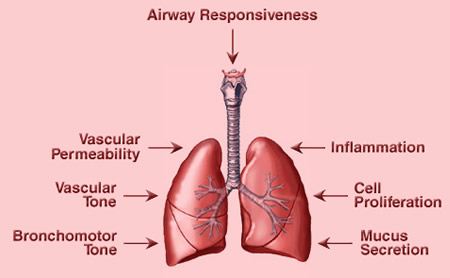
Figure 2. Effects of Prostaglandins in the Lung. Prostaglandins and thromboxane (TXA2) can regulate many lung functions. For example, TXA2 can induce bronchoconstriction, vasoconstriction and inflammation. PGE2 is generally anti-inflammatory in the lung and promotes cell proliferation and vascular permeability. PGI2 is anti-inflammatory and vasodilatory. Many prostaglandins regulate immune cell function and indirectly regulate important processes such as mucus secretion and airway responsiveness.
COX-derived eicosanoids and allergen exposure
Research in the Zeldin lab has demonstrated that airways of allergic COX-1 null mice have more eosinophils and CD3+/CD4+ lymphocytes called T helper cells (Carey et al., Am. J. Respir. Crit. Care Med., 2003). Bronchoalveolar lavage fluid (BALF) from allergic COX-1 null mice contains significantly higher levels of the Th2 cytokines IL-4, IL-5 and IL-13, increased levels of LTB4 and the cysteinyl leukotrienes, and increased levels of the chemokines TARC and eotaxin. These changes in the COX-1 null mice correlate with increased BALF IgE levels and increased MUC5AC production/mucin secretion. Moreover, the lungs of allergic COX-1 null mice exhibit increased expression of the adhesion molecules VCAM-1 and ICAM-1.
Th17 cells comprise another distinct lineage of pro-inflammatory T helper cells that are major contributors to allergic responses. The Zeldin group found that Th17 cell numbers in the lung are significantly lower in COX-2 null mice, but not COX-1 null mice, relative to WT mice following allergen sensitization/exposure in vivo. Differentiation of naïve T cells to Th17 cells in vitro requires COX-upregulation, prostaglandin production and induction of the RORγt transcription factor. RORγt induction and Th17 differentiation is diminished in COX-2 null cells but can be restored by addition of PGI2 and PGF2α. (Li et al., Am. J. Respir. Crit. Care Med., 2011). Th9 cells are known to produce IL-9 and IL-10, promote inflammation by stimulating cytokine release and leukocyte proliferation, and are involved in the pathogenesis of allergic asthma. We found that COX-2 is a key negative regulator of Th9 cell differentiation and function in the allergic mouse lung. Both PGD2 and PGE2 suppress Th9 cell differentiation through activation of their cognate DP and EP receptors, enhanced PKA signaling, and suppression of IL-17RB and PU.1 expression. Together these signaling pathways suppress IL-9 and IL-10 production (Li et al., Am. J. Respir. Crit. Care Med., 2013).
Thromboxane (TXA2) is a potent bronchial smooth muscle constrictor that is implicated in airway inflammation and hyperresponsiveness in asthmatics. Surprisingly, anti-thromboxane therapies have failed in clinical trials for treatment of asthma. In contrast to its deleterious bronchoconstrictive effects, the Zeldin group found that TXA2 attenuates Th2 and Th9 cell function in the allergic lung. By suppressing Th2 and Th9 cells, TXA2 reduced allergic inflammation and suppressed airway hyperresponsiveness. These paradoxical findings may explain the failure of anti-thromboxane treatments and suggest that systemic treatment with TXA2 mimetics may have benefit in asthmatic patients. (Li et al., JCI., 2024).
COX-derived eicosanoids and lipopolysaccharide (LPS) exposure
The Zeldin group has examined the effects of disruption of COX genes on pulmonary responses to LPS exposure. Interestingly, while COX-1 deficient mice had increased lung resistance/methacholine responsiveness following LPS exposure, there were no significant differences in inflammatory indices between the genotypes (Zeldin et al., Am. J. Resp. Cell Mol. Biol., 2001). This data indicated that in COX-1 deficient mice exposed to LPS, airway inflammation and hyperresponsiveness are distinct and that COX-1 is important in regulating physiologic, but not inflammatory, responses to LPS. Alveolar type II cells (ATII) are involved in surfactant production and recovery from lung injury. While ATII cells constitutively express high levels of COX-2, the role of COX-2 in these cells is unknown. Using mice with ATII cell-specific disruption COX-2, we found that ATII cell-derived COX-2 regulates basal airway function and LPS-induced lung inflammation, but does not play a role in bleomycin-induced fibrosis (Cheng et al., FASEB J., 2015).
COX-derived eicosanoids and bleomycin-induced fibrosis
The Zeldin group examined the role of COX-derived eicosanoids in bleomycin-induced fibrosis. Bleomycin is an effective anti-neoplastic agent, although its use is limited due to induction of lung fibrosis. Twenty-one days after bleomycin instillation, neither COX-1 null nor COX-2 null mice show changes in the extent or severity of lung fibrosis compared to WT mice. However, COX-2 null mice have significantly worse lung function compared to WT or COX-1 null mice (Card et al., Am J Respir Cell Mol Biol., 2007). Treatment of mice with either PGE2 or the prostacyclin analog iloprost is protective against the decline in lung function induced by bleomycin (Dackor et al., Am J Respir Cell Mol Biol., 2011). Together, these findings suggest an important regulatory role for COX-2 in the maintenance of lung function in the setting of fibrosis, but not in the progression of the fibrotic process.
COX-derived eicosanoids and host response to influenza virus infection
In collaboration with Dori Germolec, Ph.D., the group examined the role of COX genes in the host response to influenza virus infection, a significant cause of morbidity and mortality worldwide. The COX pathway is important in modulating immune responses and is also a major target of non-steroidal anti-inflammatory drugs. The group found that influenza A viral infection caused more severe illness in COX-1 deficient mice and less severe illness in COX-2 deficient mice; however, mortality was significantly reduced in COX-2 deficient mice (Carey et al., J. Immunol., 2005). COX-1 deficient mice had enhanced inflammation and an earlier appearance of pro-inflammatory cytokines, whereas the COX-2-deficient mice exhibited blunted inflammatory and cytokine responses along with increased viral titers. Thus, a deficiency of COX-1 and COX-2 leads to contrasting effects in the host response to influenza infection. COX-1 deficiency is detrimental, whereas COX-2 deficiency is beneficial to the host during influenza viral infection.
Future studies
In the future, we will continue to study the role of COX-2-derived eicosanoids in the pathogenesis of asthma, allergy and lung immune function. Clinical studies will determine whether COX-2 polymorphisms alter the differentiation of naïve T cells to various Th cell subsets in humans. We will also examine the role of COX-2-derived eicosanoids in regulation of γ/δ T cell differentiation and function. Our basic research on the role of COX-derived eicosanoids in modulating the pulmonary immune response to environmental agents and in the pathogenesis of asthma will provide unique opportunities for both mechanistic and translational research.