

Immunity, Inflammation, & Disease Laboratory

Research Summary
Douglas A. Bell, Ph.D., heads the Environmental Epigenomics and Disease Group and holds secondary appointments in the NIEHS Epigenetics and RNA Laboratory.
The Environmental Epigenomics and Disease Group works to characterize underlying factors that contribute to variability in human toxicological responses. We especially focus on discovery of human alleles and epigenetic factors that mediate responses to exposure and we investigate how such factors affect risk in exposed people. This basic information will be useful for understanding variability in human risk estimation models, and potentially, for identifying at-risk individuals and devising disease-prevention strategies.
Having uncovered the genetic basis for several susceptibility phenotypes in carcinogen metabolism, the group has developed high-throughput genotyping assays and worked with epidemiologists to further explore the gene-environment interaction component of exposure-induced disease. Several genotypes affecting carcinogen metabolism and DNA repair have been identified as susceptibility factors in environmentally-induced disease. The group’s gene-environment interaction studies on polymorphisms in GSTM1 (Bell et al., 1993) and N-acetyltransferase (Taylor et al., 1998) in bladder cancer have been highly cited.
The NIEHS Environmental Genome Project, the 1000 Genomes Project and Roadmap Epigenome Project have uncovered millions of sequence variants and epigenetic features in the human genome. However, relationship between these factors and transcriptional response to environmental stress is still unclear and methods for studying this are not established. The Environmental Epigenomics and Disease Group has developed novel methods that identify SNPs and DNA methylation changes in environmentally responsive enhancers (Figure 1) and have evaluated functional impact on transcription factor binding (Figure 1a, b), single cell gene expression and cellular phenotype. Thus, the group’s overall objective is to identify factors that modulate exposure responses and to evaluate their roles in human susceptibility to environmentally-induced disease using a variety of functional approaches.
Epigenetics is the nongenetic transmission of information encoded in methyl-CpGs, histone modifications or microRNAs, from parent cell to daughter cells and from one generation to the next. Epigenetic factors such as chromatin state may modulate the impact of exposure, or conversely, exposures such as tobacco smoke can directly alter epigenetic status including DNA methylation levels in enhancer regulatory sequences (Joubert et al, 2012) (Figure 2a) (Reynolds et al, 2015; Su et al, 2016; Wan et al 2018). Determining the functional impact of exposure-induced changes in methylation, such as the effect on transcription, is an active area of interest for our group (Figure 2b).
Major areas of research:
- Identification of epigenetic factors and sequence variants that modulate exposure responses regulated by the Ah receptor (carcinogen metabolism), NRF2 (oxidative stress), and p53 (DNA damage) (Figure 1).
- Evaluation of the role of these factors in environmentally-induced disease.
Current projects:
- Computational discovery and functional analysis of p53, AhR, and NRF2 transactivation target sequences (response elements) to assess the impact of SNPs on regulation of responsive genes and role in disease (Figure 1a) (Noureddine et al., 2009; Wang et al., 2011, 2016; Zeron-Medina et al., 2013; Stracquadanio G et al 2016). Understanding the role of chromatin state, dynamics, and methylation status on exposure-induced transcription of genes in the p53, AhR and NRF2 pathways (Su et al., 2014)(Figure 1b).
- Identifying exposure-induced methylation patterns in blood cell DNA (Joubert et al 2012; Su et al 2016; Wan et al 2018). A collaboration with Dr. Stephanie London (Epidemiology Branch) discovered that maternal smoking produces highly significant (p<10-15) changes in the methylation status of genes in fetal cord blood (Joubert et al., 2012). In adult smokers we see similar tobacco exposure-induced methylation patterns in their blood cells (Reynolds et al 2015; Su et al 2016) and we are determining the usefulness of these patterns as biomarkers of tobacco exposure (Figure 2a) (Reynolds et al, 2018). Using single cell sequencing and cell separation techniques combined with whole genome methylation analysis and RNA-seq we are examining the impact of exposure on hematopoietic cells from adults and neonates (Wan et al, 2018; Bergens et al, 2019) and investigating the mechanism driving these changes. A major effort is characterizing more than 30 cell types in blood using CyTof and scRNA-seq and assessing how smoking alters the transcriptome at the individual cell level. Approximately 40,000 individual cells were sequenced and the uMap clusters of cell-types are shown in Figure 3.
- In a collaboration with the CDC and NCI we are examining DNA methylation in the individuals participating in the Anniston Community Health Survey I and II (ACHS I, II) cross-sectional studies that were conducted from 2005-2007 and 2014. We hypothesize that PCBs and other persistent organic pollutant (POP) exposures in Anniston’s population may alter DNA methylation and associate with adverse health outcomes.
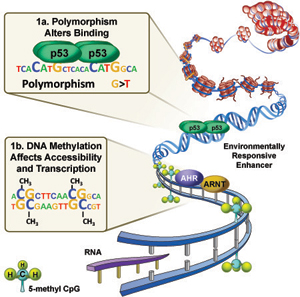
Figure 1. Environmental response genes are regulated by the binding of transcription factors sensitive to exposure signals at environmentally responsive enhancers (AhR – Aromatic hydrocarbons; NRF2- oxidative stress; p53 – DNA damage). 1A. DNA sequence polymorphism can alter binding of transcription factors to enhancers. 1B DNA methylation status can affect accessibility of transcription factors to environmental enhancers. Larger image
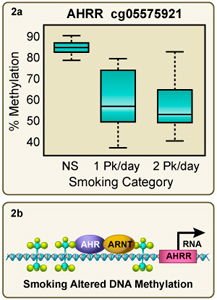
Figure 2A. Tobacco smoke exposure dramatically reduces 5-methyl cytosine levels in the AHRR gene. 2B. Reduced 5mC levels may enable AhR/ARNT binding and up-regulate AHRR gene transcription. We hypothesize that this adversely affects hematopoiesis. Larger image
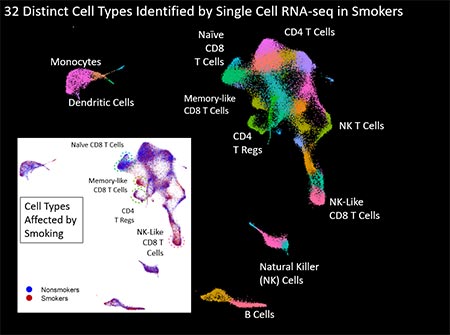
Figure 3. Figure 3 displays uMap analysis of ~40,000 blood cells collected from 4 nonsmokers and 4 smokers that were sequenced using single cell RNA-seq. We identified 32 distinct cell types (different colors) and observed significant changes in four T cell sub-types. Larger image
Bell received his Ph.D. and M.S. in environmental chemistry and biology from the University of North Carolina at Chapel Hill and a B.S. from Cornell University. He is adjunct Professor in the Department of Epidemiology, University of North Carolina at Chapel Hill. He has published over 130 peer-reviewed articles in leading biomedical journals, as well as several book chapters.
He served as Senior Editor for Biomarkers at Cancer Epidemiology Biomarkers and Prevention and has held a number of editorial board positions, including, Pharmacogenetics and Environmental Health Perspectives and has given over 80 invited lectures in the United States, Europe, Asia and South America. He served as elected chair of the Molecular Epidemiology Group, American Association of Cancer Research and president of the NIEHS faculty (Assembly of Scientists).