

Program: TAGster
Version: 1.0
September 2006
by Zongli Xu, Norman L. Kaplan, Jack A. Taylor
National Institute of Environmental Health Sciences (NIEHS)
Contents
- Legal information
- Introduction
- How it runs
- Parameters
- Output
- Conversion utility
- Usage
- Parameters
- Input files
- Requirements
- Reference
1. Legal information
TAGster License Agreement:
TAGster, including its source code and documentation, is freely distributed under the following license terms. Installation of the program on any computer or any use of the program implies that the user and the user's organization agree to the following terms:
This software is provided on an "as is" basis, with no warranty of any type, including, but not limited to, warranty of suitability for any particular purpose or ability to function correctly on any type of computer.
You may redistribute TAGster. However, the entire package, including documentation, software, this license, and source code, must be preserved.
Public Domain Notice: This is U.S. government work. Under 17 U.S.C. 105 no copyright is claimed and it may be freely distributed and copied.
2. Introduction
TAGster is a tool to select, evaluate and visualize linkage disequilibrium (LD) tag SNPs for single or multiple populations. This program analyzes patterns of LD (measured by composite linkage disequilibrium (CLD) or r2 ) between polymorphic sites in a genome region for a single or multiple populations and uses 1) a greedy algorithm to select single set containing a near-minimal number of LD tag SNPs for a single or multiple populations; 2) a computationally efficient exhaustive search algorithm to select minimal number of tag SNPs for a single population; 3) a two-stage exhaustive search algorithm to select a set of near-minimal number of tag SNPs for multiple populations; 4) a hybrid method: comprehensive use of method 1 and 3, or 1 and 2 to select tag SNPs for single or multiple populations. TAGster can also create genotype figures and LD figures with customized tracks to facilitate investigators visually checking and optimizing tag SNPs. These tracks include tag SNPs, non-synonymous SNPs, Minor Allele Frequency, tagging ability (the relative ability to capture other SNPs at a LD threshold, calculated as the number of SNPs captured by a SNP divided by the maximum number of SNPs captured by a SNP within an interested genome region) and SNP design score (probability to be successfully assayed) for each SNP.
A data format conversion utility convert is also included in order to convert HapMap or Prettybase format data to the genotype format required for use in TAGster.
3. How it Runs
On the command line, cd to the directory of TAGster and type TAGster
or double click the file TAGster in windows. On the command line typing TAGster > output_file
to output the standard screen output to the file output_file
If one wants to select tag SNPs using genotype files in HapMap format or Prettybase format, the user needs to run the conversion utility (see section 6) first, then run TAGster.
In order to produce graphical output the free software program R must be installed (see section 7).
The content of software TAGster is as following:
- 2 executable files TAGster and fconvert under root directory of tagster;
- 4 example genotype files (African, asian, ceu and hisp) under default input directory indir;
- 1 example file if the user wishes to require inclusion of certain SNPs as tag SNPs (include.txt) under default input directory indir;
- 1 example file if the user wishes to require exclusion of certain SNPs from tag SNPs (exclude.txt) under default input directory indir;
- 1 example file of a non-synonymous SNP list (nssnp) under default input directory indir;
- 1 example file of a user-specified SNP design score (score.csv) under default input directory indir; these are purely simulated values from 0 to 1, and are not actual design scores for corresponding SNPs;
- 1 perl file (snprspos.pl) to replace Seattle SNP id with rs number under default input directory indir;
- 1 example file of a list rs number and Seattle SNP correspondence (rs_example.csv) under default input directory indir;
- 6 example genotype files in HapMap format and 1 list file under default HapMap file directory indir/hapmap;
- 2 example genotype files in Prettybase format, 1 genotype list file and 1 individual list file under default Prettybase file directory indir/prettybase;
- 1 default output directory outdir.
4. Parameters
All parameters required by TAGster can be set in a self-explanatory parameter file, params.txt. The name of the parameter file “params.txt” should not be changed. In the file, lines beginning with the sign “#” are annotation lines of the immediately following parameters. For each parameter line, words or phrases before the sign “:” are the key words for that parameter. No space is allowed before key words. These key words should not be changed. User specified parameter values should be put after the colon sign. Example of a parameter file:
###lines beginning with "#" are annotation lines
###Parameter values should be given after colon ":" sign; Words before
###colon are key words and should not be changed
## run options
#Command to run R in your computer, including absolute path for R if it
is not in your system default path
-Rcommand: R
##Basic options
#task? 0:LD tag selection only; 1:selection and evaluation; 2: tag SNPs
#evaluation only
-task: 1
#selection method. 1:greedy; 2: exhaustive search for single population
or two stage exhaustive search for multi-pop; 3 hybrid;
-selection_method: 1
#Number of populations and the file names for each population data
-n_pop: 3, pop1,pop2,pop3
#directory for input file
-input_dir: indir
#directory for output
-output_dir: outdir
#LD method 1:CLD, 2:r^2
-LD_method: 2
#minimum number of informative genotype pairs required between 2 SNPs
to calculate a valid LD value between them
-Vgenopair: 5
#LD threshold
-cutoff_LD: 0.8
#maximum distance between SNPs for calculation of LD (unit: base pair)
-max_distance: 500000
##Options for tag SNP selection
#the range of SNP minor allele frequency for tag SNP selection.
#[]:include upper or lower limit; (): not include upper or lower limit
-selection_maf: [0.05,0.5]
#Minimum number of SNPs tagged by a tag SNPs, must great or equal to 1
-minimum: 1
##Options for tag SNP evaluation
#the range of SNP minor allele frequency for tag SNP evaluation.
[]:include upper or lower limit; (): not include upper or lower limit
-evaluation_maf: [0.05,0.5]<
##Options for exhaustive or two-stage exhaustive search algorithm
#max number of tries for exhaustive search
-maxtry: 1000000
##Options for additional information
#required tag SNPs list; 0: no, 1: yes; provide file name if
#yes; !!!this parameter must be set if task 2 was selected
-include_snp: 0, include.txt
#excluded tag SNPs list; 0: no, 1: yes; provide file name if yes
-exclude_snp: 0, exclude.txt
#SNP design score; 0: no, 1: yes; provide file name if yes
-score: 0, score.csv
#SNP list for non-synonymous SNPs; 0: no, 1: yes; provide file name if
#yes
-csnp: 0, nssnp
##Options for genotype or LD figures
#output figure file; 0: no figure; 1: genotype figure; 2: LD figure;
3:both figures.
-figure: 3
#sort SNP by 1: genotype similarity; 2: LD similarity; 3: chromosome
position
-sort_snp: 1
#show SNP design score track in figure; 0:no 1:yes
-track_score: 1
#show tagging power track; 0:no 1:yes
-track_tagpower: 1
#show minor allele frequency track in figure; 0:no 1:yes,
-track_maf: 1
#mark common or rare SNPs? 0:no 1:yes, followed by the cutoff value of
MAF for classification of common SNPs
-common_rare: 1,0.05
#show flags for nonsynonymous SNPs in figure; 0:no 1:yes
-track_nssnp: 1
#show major/minor allele for each SNPs; 0:no 1:yes
-track_allele: 1
#Figure type; 1: pdf; 2:eps.
-figtype: 2
The above parameter file includes a brief explanation for each parameter. A more detailed explanation is shown below.
-Rcommand
If R can not run by just type “R” in command line windows of your computer, then the parameter value here should be changed to the command to run R in your computer, including absolute path. For example “C:\Program Files\R\R-2.5.1\bin\R”. Or you could add R path into your system default path as described in Requirement section and leave this default parameter value unchanged
-task
There are 3 options for task:
0: LD tag SNP selection only; The program will select tag SNPs from SNPs with MAF specified by selection_maf.
1: Tag SNP selection and evaluation; The program will select tag SNPs from SNPs with MAF specified by selection_maf and calculate the percentage of SNPs with MAF specified by evaluation_maf captured by the set of tag SNPs at a LD threshold specified by cutoff_LD.
2: tag SNP evaluation only. The program will not select tag SNPs, instead it will calculate the percentage of SNPs with MAF specified by evaluation_maf captured by the SNPs listed in the file specified by include_snp.
-n_pop
The first parameter value is the number of populations that a user has specified, followed by file names of SNP genotype data for each population. The number of file names must equal the number of populations. If the number of populations is 1, it is equivalent to selecting LD tag SNPs from a single population. All parameter values are delimited by a comma. The program assumes each file name is a population name and therefore labels the output contents with these file names.
The genotype files follow a comma-delimited text format that contain the columns listed below.
Gene name,SNP identifier,chromosome-position,major-allele/minor-allele,SNP genotype list,MAF
For example:
Gene,snp,chr-pos,allele,id1,id2,id3,…,idn,maf
gene1,snp1,3000,A/C-1,-1,0,…,1,1,9,1,0.13
gene1,snp2,30101,C/T,0,0,…,-1,-1,9,-1,0.32
…
gene2,snp1,3040,T/G,0,-1,1,…,1,1,-1,1,0.07
…
genen,snpn,500000,A/G,-1,-1,0,…,1,0,0,0,0.43
The first line of each genotype file is a list of variable names for columns. The SNP genotype should be coded as 1, 0, -1 and 9 for homozygote common, heterozygote, homozygote rare and missing genotype, respectively.
-input_dir
This is the name of the directory that contains all input files. This directory must be created before running the program and all input files should be put in the directory.
-output_dir
This is the name of the directory that TAGster will use for all output files.
-LD_method
Specify a statistical method used to measure the LD relationship between SNPs. Parameter value 1 corresponds to composite linkage disequilibrium (CLD) and parameter value 2 corresponds to r2. If the number of valid genotype pairs between 2 SNPs (a valid genotype pair means that the genotypes for both SNPs within a single DNA sample are known) is less than 2, 0 will be assigned as the value of LD between the 2 SNPs.
-Vgenopair
A minimum number of informative genotype pairs (genotype pairs without missing data) required between 2 SNPs to calculate a valid LD value between them. If the number of informative genotype pairs is less than the specified value, a missing LD value will be assigned.
-cutoff_LD This specifies the threshold of LD for binning SNPs. It ranges from 0.0 to 1.0.
-max_distance
Maximum distance in base pair between two SNPs allowed for calculation of LD between the two SNPs. The program will not check LD relationship between SNPs if their physical distance is more than the distance specified
-selection_maf
TAGster will only use genotype data for SNPs within the specified minor allele frequency range to select tag SNPs. Use “[ ]” to include an upper or lower limit and use “( )” to exclude an upper or lower limit. The lower limit must be greater than or equal to 0 and the upper limit must be less than or equal to 0.5.
-minimum For the greedy algorithm, this defines the minimum number of SNPs required to be tagged by each tag SNP across multiple populations; for the optimal method, this defines the minimum number of SNPs required to be tagged by each population specific tag SNPs within each population. It must be greater than or equal to 1. The parameter value will substantially influence the number of tag SNPs as well as the percentage of SNPs captured by selected tag SNPs. For example, if a parameter value of 2 is set to exclude singleton tag SNPs (tag SNPs that only tag themselves in a single population during the selection process), the number of tag SNPs will be reduced by half for most genes while tagging proportions will not be decreased by much.
-evaluation_maf TAGster will calculate the percentage of SNPs within the specified minor allele frequency range captured by tag SNPs at a threshold specified by cutoff_LD. Use “[ ]” to include an upper or lower limit and use “( )” to exclude an upper or lower limit. The lower limit must be greater than or equal to 0 and the upper limit must be less than or equal to 0.5.
-maxtry For the first stage of the two-stage local optimal method in selection of population specific tag SNPs, if the number of tries in an exhaustive search is greater than the parameter value specified here, the program will switch to the greed algorithm and a warning information will be printed on a standard output. For example, “NOTE: Population xxx, Gene yyy. The number of tries 12345678 is greater than 1000000, and thus a greedy algorithm was used for the 50 SNPs and selected 10 tag SNPs”.
-include_snp Is there a list of required tag SNPs? 1=yes and 0=no. If yes, specify the file name that has a list of pre-included tag SNPs. Values of the two parameters are delimited by a comma. This parameter must be set if task 2 is selected.
The file follows a tab-delimited format that contains the columns listed below. The first row contains the variable names for columns.
Gene_name SNP_identifier
For example:
Gene Snp
gene_1 SNP_3
gene_1 SNP_7
…
gene_n SNP_k
If an investigator want to select tag SNPs using LD information from both HapMap and resequencing data, the user can use this program by first selecting tag SNPs using HapMap data, then listing HapMap tag SNPs in the include_snp file (or just copy the output file “multipop_tags.txt” to directory input_dir and specify the file name here), and then continuing on to select tag SNPs using resequencing data. This way the program will include all HapMap tag SNPs as tag SNPs and select more tag SNPs to tag the rest of SNPs in resequencing data that cannot be tagged by HapMap tag SNPs.
-exclude_snp
Is there a list of SNPs that needs to be excluded from tag SNPs? 1:yes; 0:no. If yes, specify the file name that has a list of the undesired SNPs. Values of the two parameters are delimited by a comma. The format of an exclude_snp file is the same as an include_snp file.
-score
Is there a SNP design score file? 1:yes; 0: no. If yes, specify the file name that has a list of SNP design scores. Values of the two parameters are delimited by a comma. A SNP design score can be any score that reflects the probability of successfully typing a SNP in a certain assay. The program preferentially selects SNPs with higher scores when there are multiple SNPs that can tag the same number of SNPs.
The file follows a comma-delimited format that contains the columns listed below.
gene_name, SNP_identifier,score
For example:
gene_1,SNP_3,0.387
gene_1,SNP_7,0.897
…
gene_n,SNP_k,0.656
-csnp
Is there a list of non-synonymous SNPs? 1:yes; 0: no. If yes, specify the file name that has the list of non-synonymous SNPs. Values of the two parameters are delimited by a comma. The format of a csnp file is the same as an include_snp file. This is only used for creating nsSNP track in figures.
-figure
Creating figure files? 0: no figure output; 1: output genotype figure; 2: output LD figure; 3: output both figures. SNPs with a MAF specified by evaluation_maf will be included in all figures.
-sort_snp
This parameter define how SNPs was grouped in figure. There are 3 options for grouping SNPs. Individuals will always be grouped based on genotype similarity in any options.
- Genotype similarity
- LD similarity
- Chromosome position
-track_score
Show SNP design score track in figure? 0:no; 1:yes.
-track_tagpower
Show tagging ability track in figure? 0:no; 1:yes. Relative tagging ability of each SNP (number of SNPs tagged by a SNP divided by number of SNPs tagged by the SNPs that tag the most SNPs in that gene) across all populations or within each population.
-track_maf
Show minor allele frequency track in figure? 0:no; 1:yes.
-common_rare
Mark common or rare SNPs? 0:no 1:yes; The second parameter is the MAF cutoff value for the classification of common SNPs.
-track_nssnp
Show flags for non-synonymous SNPs in figure? 0:no; 1:yes.
-track_allele
show major/minor allele for each SNPs; 0:no 1:yes
-figtype
Figure type; 1: pdf figure; 2:eps format figure.
5. Output
TAGster will print the total number of tag SNPs and a list of tagging proportions for each population into standard screen output and will also place all output files into the user’s specified output directory. Genotype and LD figures are helpful to visually check and optimize tag SNPs. Below is a more detailed explanation for each output file.
multipop_tags.txt
This is the main output file that has a list of selected tag SNPs by the program and the list of SNPs that are tagged by tag SNPs in each population. The format of the file can be found below. The first line is a list of variable names.
gene_name tag_SNP_identifier number of tagged SNPs Average_MAF Average r^2 <pop_1> tagged SNP list in population 1… <pop_n> tagged SNP list in population n
For example:
gene TagSNP Size ave.MAF ave.r2 SNPs captured
gene_1 205861 8 0.234 0.96 <pop_1> 205861 <pop_2> 205861,205190,205995 <pop_3> 205861,205995 … gene_n 168697 2 0.0545 0.91 <pop_1> <pop_2> 168697 <pop_3> 168697
untagged_snp.csv
This file lists SNPs that are not tagged by tag SNPs as listed in file multipop_tags.txt. The file follows a comma-delimited format that contains the columns listed below.
gene_name,population_name,SNP_identifier
For example:
Gene_1,pop_1,02187
Gene_1,pop_1,03243
…
Gene_n,pop_k,93456
Genotype figure
TAGster will output one genotype figure for each gene in each population. Each figure contains the following contents:
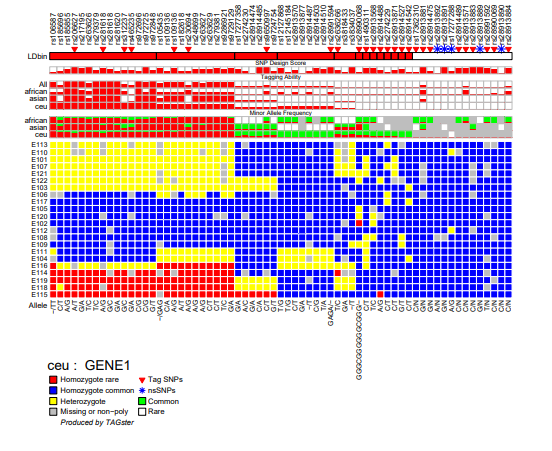
- SNP list
- Flags for tag SNPs and non-synonymous SNPs.
- Population specific LD bins (here is for CEU). The height of each bar within each LD bin is the average LD values, such as average r2 within the LD bin.
- SNP design score. The height of each bar is proportional to the probability of successfully typing a SNP in a certain assay (if this information has been included by the user in the input file).
- Relative tagging ability of each SNP (number of SNPs tagged by a SNP divided by number of SNPs tagged by the SNPs that tag the most SNPs in that gene) across all populations or within each population. Values of relative tagging power range from 0 to 1. The higher values correspond to a higher relative tagging ability. The height of each bar is proportional to the value of the relative tagging ability of a SNP.
- Minor allele frequency for each SNP within each population.
- Clustered genotype with rows referring to individuals and columns referring to SNPs
- major/minor alleles
LD figure
TAGster will output a LD figure for each gene in each population. All SNP information in the LD figure is the same as in the genotype figure. Each figure contains the following contents:

- SNP list
- Population specific LD bins and average LD values within each LD bin
- Flags for tag SNPs and non-synonymous SNPs
- SNP design score (if included by the user)
- Relative tagging power for each SNP
- Minor allele frequency for each SNP within each population
- Clustered LD
6. Conversion Utility
The conversion utility can convert SNP genotype data from Prettybase and HapMap format into the genotype format used in TAGster.
6.1 Usage:
On the command line, cd to the directory of TAGster and type fconvert
or double click the file fconvert in windows. On the command line type fconvert > output_file
to output the standard screen output to the file output_file
6.2 Parameters
All parameters can be set in the parameter file, paraconv.txt. The name of the parameter file should not be changed. The format of the file, paraconv.txt, is the same as the parameter file, params.txt
Example: paraconv.txt
###To convert prettybase or HapMap genotype format into genotype format
###used by TAGster
##Basic options
#data format; 1: HapMap; 2: prettybase;
-format: 1
#directory for output
-output_dir: indir
#maximum percentage of missing genotypes allowed for inclusion of a SNP
-max_miss: 0.9
#display program progress? 1: yes; 0: no;
-verbose: 1
## For prettybase format
#individual list and associated populations. This file should be put in
#direcoty specified by input_dir above
-ind_list: p2pid.txt
#diretory for input files
-pretty_dir: indir/prettybase
#Genotype file list; This file should be put in directory specified by
#pretty_dir above
-pretty_list: list
## For HapMap format
#only use independent individuals? 1: yes; 2: no. If yes, genotypes for
#only 60 parents in YRI or CEU will be converted. 1 was recommended.
-independent: 1
#diretory for input files
-hapmap_dir: indir/hapmap
#Genotype file list and associated population and gene list;This file
#should be put in directory specified by input_dir above
-hapmap_list: list
6.3 Input files
6.3a To convert Prettybase format
SNP Genotype data - Prettybase format:
Genotype data in Prettybase format, for example Prettybase files downloaded from seattle SNP website can be converted using this utility. Missing alleles are coded as “N”. SNP genotypes for all individuals for one gene are included in one file. It would be best if each file was named “gene_k.prettybase.txt”, replacing “gene_k” with the name of an actual gene or genome region.
Example: Prettybase format
snp1 ind1 G C
snp1 ind2 G G
…
snpn indk A C
Genotype file list
This file contains a list of of the Prettybase files with one row per file name.
Example: genotype file list
gene_1.prettybase.txt
gene_2.prettybase.txt
…
gene_n.prettybase.txt
Individual population information file
This file has the following tab delimited format: “population individual_id”. The program will use “population” specified in the file as the file names of genotype output files in the directory specified by input_dir.
Example: population information file
african D001
african D002
…
ceu E101
ceu E102
…
asian X123
asian X124
6.3b To convert HapMap format
SNP Genotype data - HapMap format
These file is one file for each gene in each population. Lines beginning with # are annotation lines.
Gene list file
This contains a list of gene names with one row for each gene in tab delimited format: population gene_name HapMap_file_name.
pop1 gene1 gene1_pop1
pop2 gene1 gene1_pop2
pop3 gene1 gene1_pop3
pop1 gene2 gene2_pop1
pop2 gene2 gene2_pop2
pop3 gene2 gene2_pop3
7. Requirements
The TAGster is written in Perl and R programming language, It can be run on Linux, Mac OS X or Microsoft Windows operating system with R installed.
The software assumes that the directory path to R has been specified in system default path and that R can be run when “R” is typed in command line. If the directory path to R has not been specified, an error message will state: ”R is not recognized as an internal or external command, operable program or batch file” in MS windows. User can either set the parameter value for “-Rcommand” in parameter file “params.txt” to the actual command for runing R from user`s computer or add the directory path to R into the system default path by doing the following:
For MS Windows XP:
- Right-click My Computer, and then click Properties
- Click the Advanced tab
- Click Environment variables
- Navigate system variables to “Path”, then click Edit
- Add the R directory to the end of variable value, such as “;C:\Program Files\R\R2.5.1\bin”. Do not forget to put a “;” to separate R path with the preceding path value.
To show system default path:
- click start
- click run
- type cmd, then click ok to open command window.
- type “path” command to show default path.
For Linux
- Type “cd” to go to home directory
- Type “vi .bash_profile” to open the system file “.bash_profile”
- Add R path to the end of the variable value for PATH, such as “:/usr/lib64/R/bin”.
Do not forget to put a “:” to separate R path with the preceding path value.
To show system default path:
- Type “echo $PATH” .
Questions can be addressed to Zongli Xu.
8. References
Zongli Xu, Norman L. Kaplan, Jack A. Taylor. TAGster: Efficient Selection of LD Tag SNPs in Single or Multiple Populations. Bioinformatics, in press (2007)