

Overview
The advent of next generation sequencing has revolutionized the manner in which DNA is sequenced and genomic events are monitored. RNA-Seq has the potential to capture the expression of every gene and its isoform(s) genome-wide. Modeling alternative tandem 3’UTRs in a dynamic fashion is an important problem in post-transcriptional regulation of mRNA and the progression of disease processes. This motivated the development of an RNA-Seq analysis method that specifically targets the 3’ UTR and dynamically models gene expression termination at polyadenylation sites. Here, RNA-Seq data and a dynamic approach are used to identify shortening of 3’UTRs. The approach uses a Poisson hidden Markov model (PHMM) to 1) estimate (hidden) states of gene expression levels in terminal exon 3’ UTRs, 2) infer shortening of the region and 3) demonstrate alternative polyadenylation.
Workflow for PHMM 3’UTR shortening
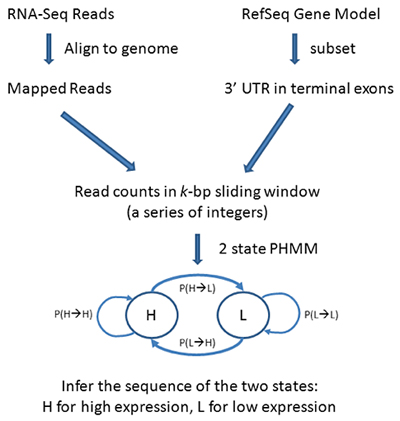
PHMM Method
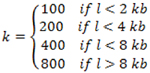
First collect all the terminal exons located within the 3’ UTR region of the gene model transcripts. A sliding window of k base-pairs (bp) is applied to each terminal exon, where the number of reads mapped to each sliding window was recorded and where:
The Poisson-based hidden Markov model (PHMM) is used to capture the sequence of read counts. In a PHMM one considers a sequence of discrete observations
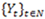
, which are assumed to be generated from a sequence of unobservable finite state Markov chains
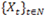
with a finite state space
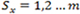
, and the random variable Yt conditioned on Xt has a Poisson distribution for every t. Specifically, if
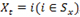
, the emission probabilities are given by a Poisson distribution with parameter

, i.e.,
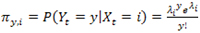
Next let Yi,j be the transition probability from state i at time t – 1 to state j at time t, and assume
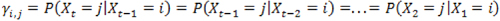
for any
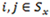
. By defining the emission and transition probabilities, along with the initial state probabilities, one can estimate all the unknown parameters for a given observed sequence  , using the maximum likelihood approach. In the RNA-Seq case,
, using the maximum likelihood approach. In the RNA-Seq case,
is a sequence of read counts with
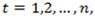
where
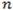
is the number of k-bp sliding windows in a 3’ terminal exon. Fit the sequence with a two-state PHMM, i.e.
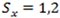
with parameters estimated by the Expectation and Maximization (EM) method. Transcripts with potential shortened 3’ UTRs (i.e. having 2 or more states) were selected based on the Bayesian information criterion (BIC) of the model fits as follows:
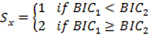
where BIC1 is the Bayesian information criterion from the 1-state model and BIC2 is the Bayesian information criterion from the 2-state model. If a 2-state model is preferred, select transcripts with transitions from high-expression state to the low-expression state (i.e.,3’UTR shortening).
Reference for Citing
Jun Lu and Pierre R. Bushel. Dynamic expression of 3’ UTRs revealed by Poisson hidden Markov modeling of RNA-Seq: implications in gene expression profiling. Gene 2013
Requirements
- R version ≥ 2.15.0
- GenomicFeatures, Rsamtools and GenomicRanges (packages from Bioconductor)
- depmixS4 (package from CRAN)
- Sorted bam file (and accompanying index file) with alignment of reads to a reference genome
Download the R Scripts
phmm.zip (7KB)
A Readme file is included to instruct on the execution of the scripts. Report bugs, corrections, and suggestions to Pierre Bushel at [email protected].
Public Domain Notice
This is U.S. government work. Under 17 U.S.C. 105 no copyright is claimed and it may be freely distributed and copied.